What is Cystic Fibrosis?
Cystic Fibrosis, often shortened to ‘CF’, is an inherited genetic condition. Over 10,000 people in the UK are affected by CF.
What causes Cystic Fibrosis?
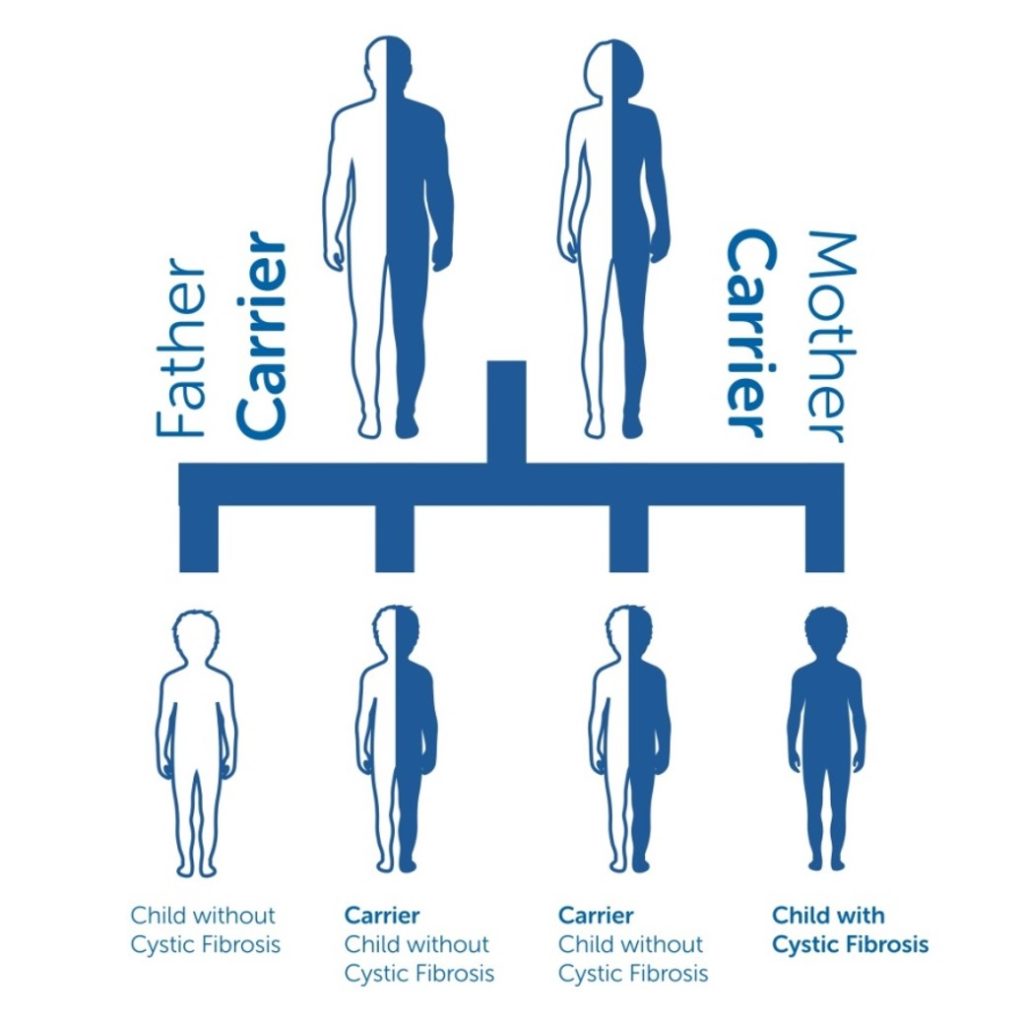
CF develops when both copies of the CF gene a person has inherited (one copy from each of the parents) is faulty. Faulty genes are called ‘mutations’. If genes are thought of as being codes for instructions to parts of the body to work in a certain way, then gene mutations can be though of as being like typing errors in that code. People with only one faulty copy of a CF gene are called ‘CF carriers.’ About 1 out of every 25 people in the UK is a carrier. Carriers are generally healthy and do not have Cystic Fibrosis. If two CF gene carriers have a baby together then there is a 1 in 4 chance that the child will inherit the faulty gene from both their mother and father and so have CF. But because there are many ‘typing errors’ causing different mutations in the CF gene, then the clinical effects of CF can be quite varied from person to person. Every person with CF is unique.
What are some common effects of Cystic Fibrosis?

The gene affected by CF is the code for the way the body controls the movement of salt and water in and out of cells. In people affected by CF this does not occur as it should, and the body’s secretions are thicker and stickier than they should be. This occurs in many organs of the body, but the effects are particularly noticeable in the lungs and in the digestive system.
The build up of sticky secretions (mucus) in the breathing tubes (bronchi) tend to predispose people with CF to coughs and chest infections. Over long periods of time this can lead to lung damage and a reduction in lung function. In the digestive system the sticky secretions tend to block the ducts in the pancreas. This means that the pancreas cannot properly secrete the enzymes needed to break down the food when eaten by people with CF. This can lead to fatty, smelly stools and to poor growth. The sticky mucus can affect other parts of the body too. Some older people with CF develop CF related diabetes and they need additional treatment to manage this.
Other problems experienced, mostly by adults, include sinusitis, nasal polyps, osteoporosis, arthritis and liver disease.
What are some of the treatments for Cystic Fibrosis?
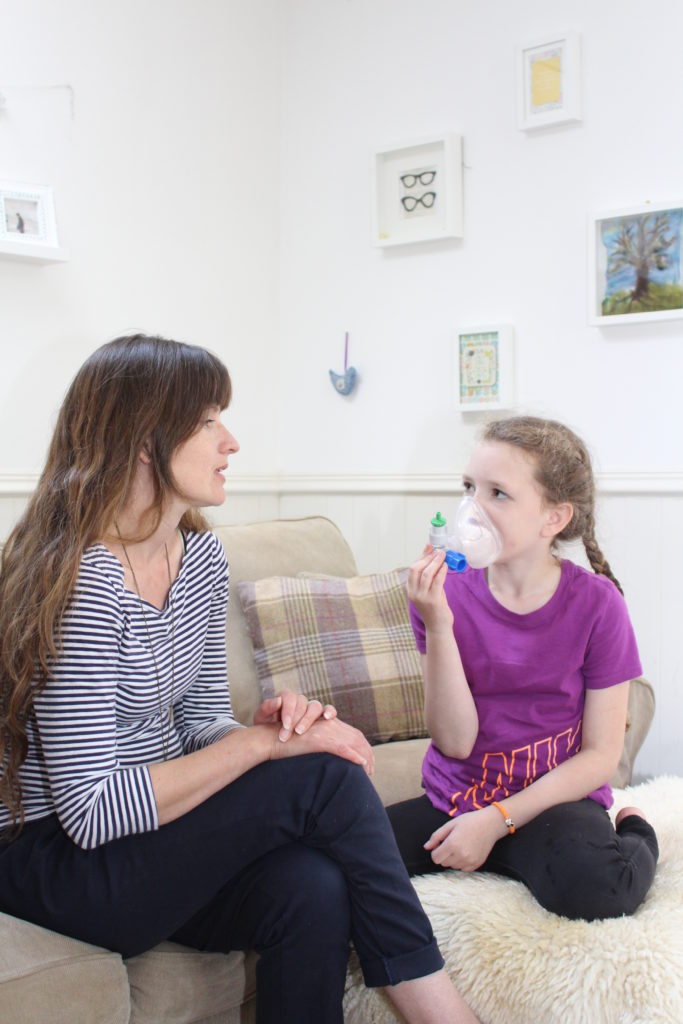
People with Cystic Fibrosis receive their health care from a multidisciplinary team of specialist doctors, nurses, physiotherapists, dieticians and other health professionals. These professionals work together to provide nationally recognised NHS standards of care. People with CF will attend regular hospital appointments to have their health monitored and the CF nurses provide outreach support to the family who provide the daily care at home.
Transitioning from paediatric care, where children and families can form attachments with the medical professionals who care for them, to adult care where the personnel and the procedures are different, can be very challenging for young people with CF. They are supported through this transition process by both teams working together for a period of time as the young person settles into the new environment and becomes familiar with the team.
While there is currently no cure for Cystic Fibrosis and whilst it is true that most people with CF will not live as long as those without CF, there are still effective treatments available, and many are carried out daily in the home.

Treatment includes:
Physiotherapy to help clear thick mucus from the lungs and help reduce chest infections and prevent lung damage.
- Parents are taught how to do physiotherapy with their child by the physiotherapist in the CF clinic. Adults with CF can learn to carry out their own physiotherapy.
- There are lots of different techniques to help clear the mucus from the lungs and the CF physiotherapist will advise and train the parents and later the young person carry out effective physiotherapy for each individual.
- The physiotherapist will also advise the parent on the frequency and duration of physiotherapy required and this may change as the person grows or when they have a bout of infection.
- Different techniques may be introduced as the person gets older.

Antibiotics and other medication to treat repeated chest infections and breathlessness. Some of these are oral medications and others are delivered by nebuliser. During periods of exacerbation of infection, some people with CF may require antibiotics delivered intravenously.
Special high fat and protein diet with vitamin supplements and pancreatic enzymes to aid digestion and prevent weight loss.
In addition to these treatments, there are many others, all of which can help specific problems that may develop. All treatment plans are designed individually around each person with CF and will differ from person to person.